

L’emofilia è una malattia genetica rara, caratterizzata da un difetto di alcuni fattori della coagulazione del sangue. In condizioni normali, la coagulazione del sangue è un processo che blocca o limita l’emorragia in caso di traumi dei vasi sanguigni. Grazie all’attivazione di numerose proteine del sangue, in una sorta di reazione a catena, nel vaso sanguigno si forma un vero e proprio “tappo”. Nelle persone affette da emofilia, due di queste proteine, il fattore VIII (emofilia A) o il fattore IX (emofilia B), sono carenti, oppure presentano un difetto di funzionamento. La conseguenza è che in caso di traumi, anche piccoli, i pazienti con emofilia hanno un elevato rischio di emorragia. L’incidenza della malattia è di 1 caso ogni 5.000 nati per l’emofilia A, il tipo più diffuso. Si registra 1 caso ogni 30.000 per l’emofilia B.
Il sintomo principale? Rischio emorragie anche per piccoli traumi
La malattia si manifesta quasi esclusivamente nei soggetti di sesso maschile, mentre le donne sono definite portatrici sane della malattia. Le manifestazioni cliniche sono simili nell’emofilia A e B. La gravità della malattia è legata al grado di carenza di attività del fattore coagulante: se il valore è minore dell’1% si parla di emofilia grave, se l’attività è compresa tra l’1 e il 5% si parla di emofilia moderata, se è compresa tra il 5 e il 40% si parla di emofilia lieve. Le manifestazioni dell’emofilia grave si osservano dalla prima infanzia. La principale è rappresentata dagli emartri, cioè versamenti articolari che si formano in seguito allo stravaso di sangue all’interno delle articolazioni, spontaneamente o per piccoli traumatismi. Altri sintomi sono gli ematomi muscolari e più raramente ematuria (sangue nelle urine), emorragie gastrointestinali, rinofaringee e cerebrali. Nei pazienti con emofilia moderata le manifestazioni emorragiche sono più rare e si verificano in seguito a piccoli traumi. Nei pazienti con emofilia lieve invece si verificano solo in occasione di traumi gravi o interventi chirurgici.
Un male “regale”
Molti dei discendenti della regina Vittoria d’Inghilterra (1819-1901) erano affetti da emofilia. Diversi discendenti maschi della monarca britannica morirono prematuramente a causa di emorragie improvvise, spesso provocate da incidenti banali come una semplice caduta per terra, prima il figlio Leopold, poi i nipoti Friedrich, Leopold e Maurice. La consuetudine dei matrimoni tra discendenti di diverse stirpi ha poi fatto sì che la malattia si diffondesse anche nelle case regnanti europee di Germania, Spagna e Russia.
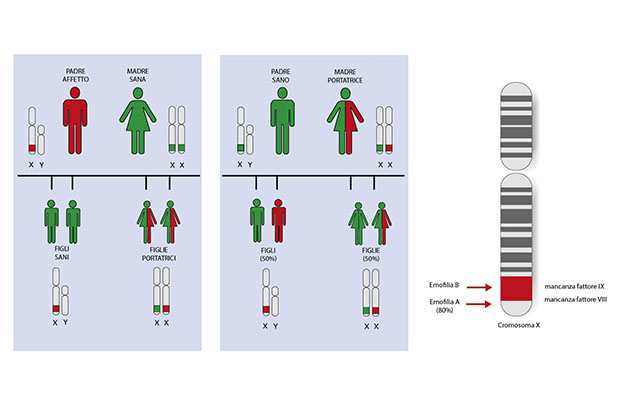
Un’anomalia ereditaria
Nella maggioranza dei casi la malattia è ereditaria. Le persone affette hanno tipicamente una storia familiare positiva per la malattia nei membri della famiglia di sesso maschile. Più raramente le anomalie a carico del gene del fattore VIII o del fattore IX si possono verificare in maniera “sporadica”. In questo caso la persona affetta rappresenta il primo caso di portatore (sano o malato, a seconda del sesso) della malattia in quella famiglia. La diagnosi si basa sull’anamnesi familiare e la storia clinica del paziente e viene confermata da test di laboratorio: test della coagulazione “aPTT” (tempo di tromboplastina parziale attivata) e dosaggio dell’attività del fattore VIII o IX.
La gestione delle complicanze
La principale complicanza è rappresentata dallo sviluppo di anticorpi “inibitori” diretti contro il fattore VIII o IX infuso a scopo terapeutico, con un meccanismo basato sul mancato riconoscimento da parte dell’organismo di tali proteine e con il conseguente sviluppo di una risposta immunitaria contro di esse. La presenza di inibitori rende inefficace la terapia sostitutiva con i concentrati di fattore VIII o IX esponendo i pazienti a un rischio aumentato di sanguinamento. Questa evenienza avviene nel 30% circa dei pazienti affetti da emofilia A grave, mentre è raro nell’emofilia A lieve/moderata e nell’emofilia B. Il trattamento consiste nell’utilizzo di “agenti bypassanti” ossia fattori della coagulazione che agiscono a valle del fattore VIII nella cascata coagulativa, come il fattore VII ricombinante o i concentrati di complesso protrombinico attivato. Sono inoltre in fase di studio nuovi promettenti farmaci biologici, anticorpi monoclonali in grado di promuovere o inibire i meccanismi rispettivamente della coagulazione o dell’anticoagulazione.
Terapie tempestive, profilassi e nuovi farmaci per una qualità di vita sempre migliore
Per quanto riguarda l’emofilia A grave, la cura si basa sulla somministrazione di concentrati di fattore VIII: in profilassi (a giorni alterni o tre volte alla settimana), oppure al bisogno, in occasione di un evento emorragico o in preparazione a interventi chirurgici. La profilassi primaria si inizia entro i primi tre anni di vita, prima che si sviluppi l’emartro. Quando la profilassi viene iniziata dopo 1-2 episodi di versamento articolare, ma prima che si sviluppi il danno all’articolazione, è detta secondaria. È terziaria quando viene iniziata quando già presente il danno, un evento fortunatamente ormai poco frequente. Il trattamento è analogo nell’emofilia B, che si basa sulla somministrazione di concentrati di fattore IX. Per i pazienti con emofilia lieve o moderata è sufficiente un trattamento al bisogno. La profilassi ha radicalmente modificato la storia naturale della malattia, consentendo di prevenire le gravi alterazioni dell’apparato muscolo-scheletrico, principale causa di morbilità e invalidità nel paziente emofilico adulto e causa di notevole compromissione della qualità della vita. Per migliorare ulteriormente la qualità di vita dei pazienti, sono stati sviluppati e da poco messi in commercio farmaci “a emivita prolungata”, con lo scopo di fornire una protezione prolungata dalle emorragie, riducendo la frequenza delle infusioni. Per il futuro ci si potrà avvalere della cosiddetta “terapia genica”, cioè la sostituzione del gene malato con un gene sano. Gli studi sono in fase avanzata per l’emofilia A, mentre sono più problematici per l’emofilia B.
A cura della Dott.ssa Anna Falanga
Direttore del Dipartimento interaziendale Medicina Trasfusionale ed Ematologia della provincia di Bergamo (DMTE) e del Servizio di Immunoematologia e Medicina Trasfusionale (SIMT)
ASST Papa Giovanni XXIII